Communications of the ACM
Deep Learning Aces Protein Folding

DeepMind's AlphaFold 2 is able to predict the three-dimensional shape into which specific chains of amino acids will fold.
Credit: DeepMind
London-based DeepMind is renowned for using deep learning to beat humanity's best Go player. In December, however, the organization revealed its prowess against Mother Nature by predicting the three-dimensional shape into which specific chains of amino acids will fold.
Recognizing the biological and medical significance of the folding question, researchers have a longstanding project to compare different algorithms. This biennial "Critical Assessment of protein Structure Prediction," or CASP, has documented steady progress over the last 26 years. DeepMind's entry, AlphaFold 2, blew the field away.
"They pretty much got it," said Andrei Lupas of the Max Planck Institute for Developmental Biology in Tübingen, Germany, who volunteers as an "assessor" for CASP. "It was the kind of breakthrough that changes the game completely."
Proteins are long molecules that string together scores or hundreds of the 20-odd amino acids. Their precise order is specified by the genetic information in DNA, which is now readily and cheaply determined. To understand protein function, however, researchers need to know how this chain folds into a compact molecular machine, choosing one configuration from among a vast number of possibilities (historically estimated as 10300).
Protein structure was historically derived from x-ray scattering, which requires painstaking preparation of large, purified crystals, or from nuclear magnetic resonance. More recently the field has benefited from electron microscope characterization of smaller flash-frozen samples.
These experiments have revealed important organizational principles, such as attractions between various types of amino acid, as well as recurring structural motifs like helices and sheets. Predicting which overall configuration has the lowest energy from the sequence alone, however, has remained a daunting computational task.
Blinded Assessment
Early on, there were concerns that researchers might be tweaking their programs, perhaps unintentionally, to better match the measured protein structure. Started in 1994, CASP avoided this problem by announcing the sequences of proteins but withholding the structure until after the predictions are finished. Specialists like Lupas prepare these target tasks and evaluate the results every two years.
The level of agreement is regularly assessed using a metric called the Global Distance Test (GDT, specifically the "TS" or total-score variant), which is the percentage of amino acids whose predicted positions are closer than some threshold to the experimental value. For a decade, CASP participants have struggled to beat a median score of 40 in the challenging free-modeling category.
DeepMind first entered CASP in 2018, and their AlphaFold tool achieved a significantly improved GDT of 60. For the 2020 assessment, they completely modified their architecture to use attention-based methods, which have excelled in large-scale language models. This approach, building up from low-level details, differs from most previous approaches that began with a top-down view of the complete structure. The team also used end-to-end training of the model, reinforcing good structure predictions from starting sequences, rather than monitoring intermediate steps. For training, they exploited databases including some 170,000 protein-structure/sequence combinations, as well as other data.
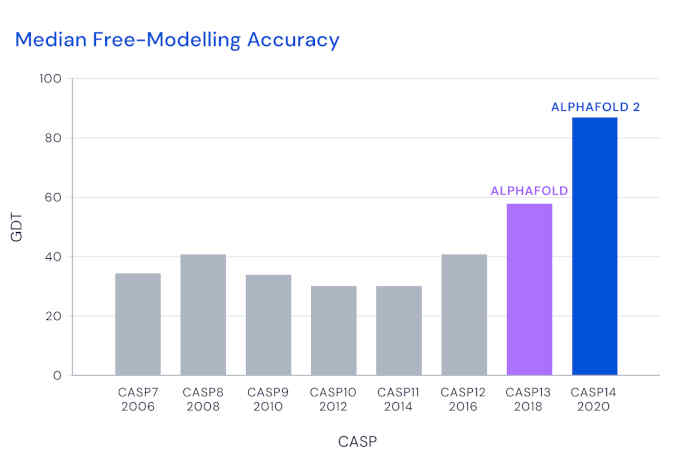
With these methods, AlphaFold 2 achieved a GDT of almost 90 in the latest CASP, with each structure requiring just a few days of computation. This enormous improvement reflects an accuracy roughly comparable to the experimental uncertainty. Lupas and his colleagues also "worried that they were doing just too well," so they included a protein for which they had struggled to find a structure for a decade, despite having excellent x-ray data. He said that "with their model, the structure fell right out within a few minutes."
DeepMind declined an interview for this story, but after its 2018 success, the organization later published many of the technical details. Although "not enough details have been provided" so far, Lupas said, "AlphaFold has been plenty open with us, telling us what the key insights were." As a result, "Everybody is now playing with attention nodes. Most people are doing end-to-end networks," he said. "There's a lot graduate students who are not sleeping much."
Putting Prediction to Work
"As more and more accurate structure predictions become available, it will really help with illuminating biology that you previously would have needed experimental structural determination for," said David Baker of the University of Washington, whose Rosetta tool was a leader in previous CASP assessments. Still, he noted, "Usually it's not just the structure, but experiments also on top of that structure, that are needed to get biological insights."
Nonetheless, AlphaFold and similar prediction tools can be generalized to illuminate biological function. For example, proteins frequently bind to each other in pairs or larger complexes, or to DNA or RNA, to perform specific biological functions. The spatial interactions of proteins with small molecules can also show how they promote chemical reactions, or how those small molecules might serve as drugs to modify their actions.
One outstanding question, Lupas noted, concerns the many proteins, or regions of proteins, whose structures are not precisely predetermined. "Everything in CASP has a structure. That's one of the premises," he said. He hopes to learn if AlphaFold or its successors can tell whether a particular structure is unique. "Does this network know when there is no solution?"
Baker's team has developed principles for designing proteins that have no natural analog, several of which are in clinical trials. In work modeled on the earlier version of AlphaFold, "It actually did a remarkably good job at predicting the structure of our de novo designed proteins," Baker said. "These networks are clearly learning general principles," just not in a form that people can easily understand.
Don Monroe is a science and technology writer based in Boston, MA, USA.
No entries found